

Alice Lee, Anna Sun, Ayden Maza, Kimberly Stinson, and Simon Han
Vol. IV, No. I
I. Introduction
The bureaucratic nature of America’s healthcare system creates an overly complex regulatory framework that comes at the expense of consumer ease and care. The exorbitant prices for life-saving medications are controlled and manipulated by corporations with a vested economic interest in the healthcare industry, leaving citizens at the mercy of powerful regulation agencies and influential companies. The current state of the healthcare industry, with respect to food and product safety, will be explored here through an overview of the history that has informed regulatory practices today and the complicated landscape of contemporary consumer health regulation. This article aims to evaluate food and product safety as a public health concern through a socio-legal analysis of three primary stakeholder groups: government regulatory agencies, private industry, and individual citizens. By examining the ways in which various public and private actors implement the laws, enforce the laws, circumvent the laws, and live by the laws, this article seeks to address the structural challenges within the United States food safety system and the unequal power dynamics that continue to reinforce its problematic elements.
II. History of Food Safety in the United States
Since the beginning of the twentieth century, the concept of consumer protection has been shaped by public outrage. One such area of concern was the meatpacking industry and its unsanitary working conditions, as revealed by Upton Sinclair in The Jungle. [1] Spurred by the public health necessity to remedy the dangerous nature of the food industry, the first comprehensive inspection authority was created in 1906—the Food and Drug Administration (FDA). President Roosevelt’s signing of the Pure Food and Drug Act established the federal government’s responsibility for consumer protection. Though the 1906 law set forth the first legal standards for food and medicine protection, regulations regarding misleading ingredient or statement information in advertising and provisions for food quality were excluded. [2] Over the course of the twentieth century and the ensuing Progressive Era, state, local, and federal agencies implemented more clearly defined jurisdiction and guidelines to goods and services within the scope of consumer protection laws. The gradual development of these policies ran parallel to a heightened emphasis on safeguarding consumers from unsafe products, unfair business practices, and fraud or deception. However, these protective measures lack centralization in that they are quite varied and exist separately.
In the United States, food safety regulation is dispersed among a multitude of bureaucracies that each individually handle various elements of the overall process. Food safety refers to the proper handling, storing, and preparing of food to prevent infection or contamination and to ensure the quality of food is preserved. Government regulation of factory conditions and practices is key to protect food safety. From product inspection to consumer education and compiling disease outbreak statistics, regulatory responsibilities are divided and shared by agencies at the federal, state, and local levels. Of the fifteen agencies legally mandated to ensure food safety, the Food and Drug Administration (FDA), the U.S. Department of Agriculture (USDA), and the Centers for Disease Control (CDC) play arguably the largest roles. [3] The majority of the oversight lies within these three federal organizations. The USDA ensures that the country’s meat, poultry, and processed egg supply (both domestic and imported) is safe and properly labeled; the FDA is responsible for the rest of the domestic and imported food as well as animal feed and drugs; and the CDC addresses foodborne illness and disease outbreaks.
The FDA protects public health by ensuring the safety and efficacy of an extensive range of food- and health-related products, including cosmetics; therefore, it is a critical part of the U.S. public health infrastructure. The FDA is charged with providing oversight for “protecting the public health by assuring the safety, efficacy, and security of human and veterinary drugs, biological products, medical devices, our nation’s food supply, cosmetics, and products that emit radiation.” [4] Over the last century, the scope of the FDA has increased substantially—as have class action lawsuits filed by plaintiffs who seek remedy from food giants for deceptive marketing practices that fail to be regulated by federal agencies. The “unprecedented surge” in consumer lawsuits against the manufacturers of products such as Naked Juice, Fruit Roll-Ups, and Wesson Oil reveals a trend of regulation by litigation—turning to the courts to address food labeling issues because of a lax regulatory system. [5] The FDA lacks the resources and regulatory authority to effectively monitor false or misleading labeling practices, resulting in consumer confusion and an uneven playing field in the health marketplace.
III. FDA Regulation of Drugs & Tobacco
Further criticisms of the FDA specifically “center on the extent of its regulatory powers and the risk that FDA officials could be swayed by the powerful pharmaceutical lobby,” arguing that the agency should have more power and independence to prevent such influence from prominent actors, like those in the pharmaceutical industry. [6] This section addresses the role of the FDA in regulating prescription drugs and tobacco products through a historical investigation of court cases which pitted the interests of Big Pharma against the government’s responsibility to safeguard public health.
A. OxyContin
The FDA has one of the most important roles in public health: to regulate the pharmaceutical industry. This is quite a significant role to conduct since some medications can have dire health consequences if allowed to enter the market. The FDA faces, arguably, a large problem with regulating major pharmaceutical companies, or Big Pharma. Big Pharma holds a strong influence over the pharmaceutical industry by controlling research, production, and advertising in the healthcare market. One of the largest issues the FDA has faced is the opioid epidemic. In 1995, the FDA approved the use of OxyContin, a strong prescription pain medicine that contains an opioid (narcotic). However, in 2007, Purdue Pharma, the company that owns OxyContin, was found guilty of lying to the public because they withheld information about the addictive nature of the drug. [7] During congressional hearings before the end of the trial, key information was brought to light that made a convincing argument as to how OxyContin was the catalyst for the opioid epidemic.
Prior to the approval of OxyContin, the FDA and Purdue Pharma were aware that a delayed reaction could be counteracted simply by crushing the tablet. [8] Although data at the time showed no signs of abuse with time-release MS-Contin, the FDA thought that simply putting a warning about not crushing the tablet before ingestion would prevent people from doing it. A simple fix that could have reduced the prevalence of OxyContin abuse and addiction would have been to require Purdue Pharma to fix the flaw in their product before approving the dangerous drug for public consumption. Yet the FDA took no action to regulate the health concern and permitted OxyContin to enter the market. Another key factor in the spread of OxyContin usage was the heavy advertising and promotion by Purdue Pharma. By training sales representatives to lie about the drug’s addictive potential, Purdue Pharma misrepresented OxyContin as non-addictive and even unlikely to lead to dependence and abuse.
However, in U.S. v. Purdue Frederick Co., the district court found that Purdue Pharma did have knowledge that their drug could become addictive and lead to abuse. [9] Again, the lack of action the FDA took in regulating Purdue Pharma’s deceptive marketing strategies and its failure to intervene when it knew there was a flaw in the drug was a major mishap. To sum up the actions of the FDA and Purdue Pharma: “regulation deficiency resulting in state-corporate victimization.” [10] The FDA had good intentions to protect consumers from the drug, yet the lack of policy at the time and enforcement of their policies helped add fuel to the flame for the opioid epidemic. Unwillingness to force Purdue Pharma to change the structure of the pill before approval is a clear example of regulatory deficiency. The insufficient oversight is further illustrated in the fact that the FDA does not directly conduct any tests on drugs. [11] A change could be made in which the FDA performs its own testing procedure instead of relying solely on pharmaceutical companies’ data. Bolstering the FDA’s role with real and substantive power would allow for safer and more informed decisions about drugs before they reach the market.
B. Tobacco
When looking into the effectiveness of FDA regulations over time, the tobacco industry is one of the best to investigate. The FDA and the federal government face many issues when it comes to regulation of tobacco companies, one of which is honesty about the product. In 1994, the CEOs of tobacco companies testified before Congress about tobacco products and their health effects for the first time in U.S. history. During their testimony, all seven of the CEOs present testified that nicotine was not an addictive drug. [12] For the tobacco industry to be under oath in front of Congress and not even admit to the fact that their products are addictive is a major problem for regulators. Given the ease with which CEOs can lie to Congress about the effects of tobacco, it is clear that companies can shape the public narrative to favor themselves—a coercive element of industry that restricts the ability of the government to regulate properly. Without all the facts, the government cannot do its job of regulation.
In 1996, the FDA attempted to regulate the tobacco industry under the Food Drugs and Cosmetics Act (FDCA) by stipulating that tobacco products fall under the “drug” and “devices” definition that allows the FDA to regulate. [13] However, tobacco companies filed a lawsuit to challenge the regulation and won. In FDA v. Brown & Williamson, the Supreme Court ruled that Congress had not granted the FDA the power to regulate tobacco. [14] The Court also found that Congress had rejected proposals to give the FDA this authority and acted to prevent this authority from being granted. Up to this point in time, no progress had been made in public health to regulate the danger of tobacco products.
Eventually, these companies were found guilty of lying in a major lawsuit between the federal government allied with anti-tobacco foundations and tobacco company Philip Morris USA in 2006. In the decision written by Justice Gladys Kessler, it was determined that tobacco companies purposely lied in advertising tobacco products and hid the addictive properties and health impacts of the product. [15] From this decision, tobacco companies now had to include disclaimers on their products about the addictive potential of tobacco, the effects of secondhand smoke, and even that smoking “low tar” or “light” products does not have many health benefits. With more information revealed about the harmful consequences of tobacco, the FDA gained the ability to regulate the manufacturing, distribution, and marketing of tobacco products in 2009 with the passage of the Tobacco Control Act. [16] Now, in a new age of smoking, the FDA can regulate e-cigarettes, as decided in the ruling of Sottera, Inc. v. FDA. In this case, the Court granted the FDA the ability to regulate any product made or derived from tobacco. [17] Overall, it took time and extensive resources to establish the regulations we see today from the FDA on tobacco products. This history suggests that with support from Congress, proper regulations can be imposed on companies to help protect the public.
C. Insulin
The regulation and accessibility of insulin in the United States has been a complex issue for quite some time. The reason for this is that three multinational companies control 99% of the insulin market: Eli Lilly, Novo Nordisk, and Sanofi. [18] With three companies dominating the market, it has become hard for any type of competition to curb the monopoly in the insulin market, a trend which has allowed these companies to set unreasonably high prices. Without regulatory policy to combat this monopolization, it is extremely hard to envision greater access to affordable medication in the United States. Nevertheless, the FDA has continued to attempt to increase access to insulin and other drugs on the market. In 2017, the Drug Competition Action Plan was introduced by the FDA to help promote competition through generic drugs, making the approval process easier and even closing some loopholes in the system that block generic drugs from being approved. [19]
Further, the FDA took a stand against companies when Congress mandated that the FDA change the regulatory pathway for insulin to allow for biosimilar products. [20] By opening the way for biosimilar products, generic insulin could finally become an option for Americans. These new regulations are designed to help decrease the power of the “Big Three” companies and allow for more people to gain access to insulin. On top of these regulations, Illinois Public Act 101-0625, passed in 2020, changed the state insurance code to cap insulin costs to $100 a month. [21] A similar bill was passed in the House of Representatives that caps cost-sharing to $35 a month. [22] With these steps taken by the government, new layers of regulation could help protect consumers, eventually allowing for less abuse from pharmaceutical companies and greater access to medication for consumers.
D. Analysis
Valuable information can be gained from studying FDA and federal government responses to situations in the public health industry. A common thread that holds true in all three areas (OxyContin, tobacco, and insulin) is “regulation deficiency resulting in state-corporate victimization.” [23] The ability of pharmaceutical companies to get away with their actions primarily stems from the FDA and government lacking the forceful policies that are needed to restrict companies’ actions. This lack of regulation has allowed these companies to do whatever they want; since no regulation or law stops them, they remain in legal standing. On top of this, the regulations currently in place do not adequately ensure consumer safety. Drug companies are allowed to advertise as much as they want and are only required to briefly bring up the side effects of their drugs. The lack of resources all comes back to how the government has not allowed the FDA to do the job it can. One sign that this is true is how approximately 45% of the FDA’s budget comes from user fees that drug companies pay. [24] In other words, the companies that are supposed to be getting regulated are funding almost half of the FDA. It seems evident that an innovative approach to food and drug regulation is needed, one in which the federal government takes a more active role in medicine and regulations against companies. Specifically, the federal government should continue to give the FDA more regulatory power, as it has done in recent years.
IV. International Comparison
One of the largest differences between the United States and other countries in the world is the healthcare system that they have. For starters, the United States differs from most Western industrialized countries in that it does not have a centralized, single payer healthcare system. The United States runs on private healthcare for Americans along with federally-administered Medicaid and Medicare. Medicaid provides healthcare for low-income Americans and varies from state to state. [25] Medicare is health insurance for Americans 65 or older and certain young people with disabilities. [26] One main factor that is apparent in the United States is the lack of policy in healthcare that helps Americans. Unless you fall under a certain status, you are left to find private healthcare on your own. However, most other advanced and developed nations have more government activity in health services. Germany, serving as the largest economy in Europe, has a mixed system with centralized health insurance and private health insurance. In Germany, citizens earning less than $35,000 per year qualify for sickness funds, while higher earners can choose a private insurance option. [27] This system allows every citizen in Germany access to healthcare, regardless of income level. Thus, the German healthcare system is quite accessible since it is required by law to have either public health insurance or a private version.
On top of this, most countries in Europe also engage in drug price negotiation. In Germany, a scientific entity overseen by the German Ministry of Health negotiates the drug prices for one universal value for the sickness fund. [28] With the negotiating, the drug prices are lower compared to the prices in the United States: “The drug prices set through these combined approaches are significantly lower than those in the U.S., and the gap appears to be widening over time. U.S. net prices for the most expensive drugs are up to four times higher than their German equivalents.” [29] The methods used in other wealthy, industrialized countries like Germany help show how more people are enabled to be ensured and even how prices for the same drug are lower with simply more intervention from the government. If the United States were to take a more hands-on approach to healthcare, there would be less of a burden on the American people. And the approach of more hands-on healthcare can be seen to work in countries similar to the United States, since every Western developed nation has some form of government healthcare to help patients. The United States ought to follow this well-established model and grant the federal government a more active role in healthcare.
V. FDA Regulation of Cosmetics
This section examines the relationship between personal care product regulation and resulting lawsuits, industry players, and Congress. Asbestos findings in trusted consumer giant Johnson & Johnson’s products and the thousands of lawsuits resulting from the lack of industry regulation highlight the problems stemming from an absence of adequate oversight. Further, reform of the FDCA along with the passage of other transformative legislation will be paramount to strengthening the Food and Drug Administration’s regulatory authority, as will providing more stringent industry oversight with the goal of adopting a precautionary approach for ingredients in cosmetics to protect the livelihood of future generations.
Within the last decade, members of Congress have begun to devote their efforts toward consumer safety as more research comes to light revealing adverse health effects of harmful ingredients used in consumer products. As more advocacy and education about health impacts increase, concerns from consumers and professionals in the industry grow regarding personal care products and the detrimental impacts they may have on susceptible persons. Susceptibility to harm often stems from a lack of information available on product ingredients, the number of chemicals and formulations in them that have not undergone toxicity testing, the unknown health impacts for long-term low-dose exposure to individual chemicals or chemical mixtures, and insufficient consumer standards and enforcement. [30]
Congress passed the FDCA in 1938—over 83 years ago. Since then, the cosmetics industry has grown to be a multibillion-dollar industry “with products being marketed worldwide and sold not only in retail stores but by individuals working out of their homes and over the Internet.” [31] Regulatory quality captures perceptions of the ability of the government to formulate and implement sound policies and regulations that permit and promote private sector development. From the first major federal law in 1938 to present-day regulations, the rules governing the FDA and personal care products have evolved over the years; still, the clean beauty movement calls for further reforms to protect consumers in the years to come.
The Food, Drug, and Cosmetic Act is one of the most important laws pertaining to cosmetics in the U.S. The Act defines the term “cosmetics” as “articles intended to be rubbed, poured, sprinkled, or sprayed on, introduced into, or otherwise applied to the human body… for cleansing, beautifying, promoting attractiveness, or altering the appearance.” [32] Companies and individuals who manufacture or market cosmetics have a legal responsibility to ensure the safety of their products. However, neither the law nor FDA regulations require “specific tests to demonstrate the safety of individual products or ingredients. The law [FDCA] also does not require cosmetic companies to share their safety information with [the] FDA.” [33] Only a few regulations prohibit or restrict the use of several ingredients in cosmetic products and require warning labels.
A. Johnson & Johnson
The lack of regulation and adequate testing of talc-containing personal care products in the U.S. has led to the contamination of cosmetics with asbestos. Talc, a mineral ingredient, has been used in personal care products “for decades,” but is also “consistently found to be contaminated with amphibole asbestos.” [34] Thus, Stoiber et al. allege that “the true exposure of consumers to asbestos is poorly characterized and likely underestimated.” [35] Stoiber et al. additionally highlight the ineffectiveness of current voluntary screening methods and investigate the occurrence of asbestos in talc-containing cosmetics products by evaluating 21 talc-based cosmetics products, finding that three of the 21 products tested were contaminated with asbestos. [36] Their results successfully demonstrate the necessary urgency to revise cosmetics policy.
Johnson & Johnson (J&J) is the world’s largest health care company and the highest-paid drug company globally. [37] Among its most popular consumer brands is Johnson’s Baby Powder. J&J “has been at the center of scandals and government investigations over the years” and faces an increasing share of lawsuits. [38] In October 2019, due to the FDA findings of asbestos in a product sample and “out of an abundance of caution,” J&J bowed to increasing public pressure and recalled approximately 33,00 bottles of its Johnson’s Baby Powder. [39] The FDA lacks the authority to recall cosmetics, as cosmetic recalls are voluntary actions taken by manufacturers or distributors. However, the agency “monitor[s] companies that conduct a product recall and may request a product recall if the firm is not willing to remove dangerous products from the market.” Moreover, the FDA “has the responsibility to monitor the cosmetics market and can take appropriate action to protect consumers in the post-market setting.” [40]
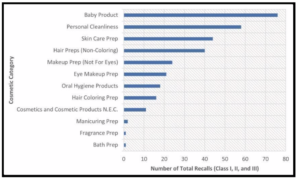
A 2018 study by Janetos et al. published in the Journal of Cosmetic Dermatology describes all FDA cosmetic and personal care product recalls, acquired via a Freedom of Information Act (FOIA) request, from 2002 to 2016. [41] The total number of Class I, Class II, and Class III recalls, number and origin of manufacturers, number of products affected, and foremost reason for recall were collected. As the authors state, “During the study period, a total of 14 Class I, 266 Class II, and 33 Class III recalls were recorded. Baby products comprised the largest product category accounting for 24% of all recalls (76/313), followed by personal cleanliness products.” [42] The 14 Class I recalls included over 1.9 million products, the 266 Class II recalls included over 23 million products, and the 33 Class III recalls accounted for over 2.7 million products in distribution at the time of recall. [43] The study concludes that “cosmetics recalls impacted millions of products and had the potential to cause harm.” [44] Furthermore, Janetos et al. assert that “dermatologists have the ability to strengthen public safety by reporting adverse events, encouraging recalls of harmful products, and lobbying through dermatology organizations for meaningful change to current cosmetic regulation.” [45]
B. Findings
Voices about cosmetic and personal care reform were relatively quiet until March 2012, when the House Energy and Commerce Subcommittee on Health held a hearing entitled “Examining the Current State of Cosmetics.” [46] One of the witnesses was Michael DiBartolomeis, Chief of the Safe Cosmetics Program in the California Department of Public Health—the first state cosmetics-regulatory program in the nation. His testimony addressed some of the main concerns he had heard from numerous consumers and professionals in the personal care industry. DiBartolomeis stated in his testimony:
“While the industry has changed, the provisions of federal law for regulating cosmetics have not. As a result, the burden falls on government to show harm before a cosmetic can be removed from the market. No premarket safety testing is required. Manufacturers have almost no incentive to test products for their potential to cause serious latent harm such as cancer. Cosmetic labels are not required to disclose some ingredients, and there are no requirements to disclose them to the Federal Government. And chemicals that can cause cancer, reproductive and/or developmental harm are consistently ending up in cosmetic products” (italics ours). [47]
By emphasizing the inadequacy or outright absence of toxicity testing and reference to exposure to chemicals, Dr. DiBartolomeis’ testimony represents consumer and industry concerns exacerbated by the asbestos findings in J&J’s Baby Powder.
In Pace Environmental Law Review, Katherine Drabiak focuses on the link between J&J’s talc-based product and health risks (e.g., ovarian cancer) required by courts for success in these lawsuits. She emphasizes the harms of endocrine-disrupting chemicals (“EDCs”) in chemical-containing cosmetics products. [48] Drabiak uses Hogans v. Johnson & Johnson (2014) to illustrate that the current risk-based regulatory system fails to ensure consumer safety from toxins. [49] She describes how routine cosmetic products contribute to increased cancer risk, presenting a public health concern. Toxicants in personal care products are “numerous, scientifically troubling, and present in even the smallest of members of the population.” [50] Along with this, much of the public is oblivious to the FDA’s “lack of stringency pertaining to cosmetics and the risk of their toxic ingredients.” Drabiak asserts a correlation between the increase in cancer and the increase in synthetic chemicals like those in cosmetics. [51]
C. Analysis
The FDA’s resources and authority to regulate the cosmetic industry are limited. The agency possesses no power to ensure the pre-market safety of cosmetic products or force companies to pull them off the shelves when potential hazards are discovered. Without regulation, manufacturers in the United States can formulate and sell products containing chemicals that are carcinogenic, mutagenic, or toxic for reproduction.
The danger of J&J’s Baby Powder exemplifies the importance of bills that propose increases in regulation and safety of cosmetics and other products outside of the personal care product industry. There are many ongoing efforts to reform the FDCA. One bill, authored by Congressman Frank Pallone, the Cosmetics Safety Enhancement Act of 2019, would “require cosmetics companies to substantiate the safety of their products, notify the Food and Drug Administration of any adverse health events, give the FDA the power to conduct its own safety reviews, and mandate that manufacturers provide more transparency about ingredients on their labels.” [52] In addition, the FDA Cosmetic Safety and Modernization Act and the Personal Care Products Safety Act are two other significant bills designed to address the lack of regulation in the cosmetic industry.
Mass-tort lawsuits, congressional hearings and investigations, reactions from various industry actors, and actions (or lack thereof) taken by the FDA illustrate the need for further FDA regulation of personal care products, which will only be possible through the passage of new laws authorizing enforcement of higher standards for cosmetics. Although “the FDA’s risk-based approach prohibits manufacturers from placing adulterated or misbranded products in the stream of commerce, the FDCA does not require the FDA to assess the safety of the ingredients of cosmetics, and the FDA has no authority to recall demonstrably harmful cosmetics.” [53]
Currently, the FDA can only act if a cosmetic is adulterated or misbranded, despite findings which suggest that chemicals used in many products cause long-term harm when used in cosmetics. However, the current structure of the FDCA does not enable the FDA to evaluate these potential harms due to the limited information the FDA has on cosmetic ingredients and the current focus on reacting to complaints from consumers in the marketplace. [54] Congress must pass new legislation giving the FDA broader authority to regulate harmful ingredients in cosmetics. Recently proposed reforms, such as the Personal Care Products Safety Act, would improve reporting by making registration mandatory and would put the FDA in a better position to review the safety of cosmetics across the market. More substantive changes such as allowing the FDA to set limits on ingredients below the safety threshold based on a precautionary approach may also be necessary to deal with the cumulative effect of consumer exposure. [55]
Advocates point to the European model of personal care product regulation as an avenue for reform in the United States. Compared to the 1,328 prohibited ingredients in the European Union (EU), the FDA has only banned or restricted 11 chemicals from cosmetics. [56] Wallack adds that the EU “does not technically require products to be pre-approved, but it does mandate that manufacturers conduct a much more stringent safety assessment than does the United States.” [57] In addition, “the EU requires all products to be registered in the Cosmetic Products Notification Portal–an EU-wide database–before they can be marketed in the EU.” [58] Because the EU safety assessment focuses more on systemic harms from use of the product than on individual ingredients, it may be better suited to dealing with the problem of repeated, long-term use. [59]
A long road lies ahead in the fight to pass new legislation governing FDA regulation of cosmetics and personal care products. Aside from the introduction of new bills like the Personal Care Products Safety Act, “while the industry has changed, the provisions in the federal law for regulating cosmetics have not.” [60] The clean beauty movement is not just about lipstick; it is about the safety of the public and the need for new personal care product regulations to protect the livelihood of future generations.
VI. History of Corporate Rights
This section will evaluate the role and influence of corporations on public health regulation and enforcement. The Fourteenth Amendment of the U.S. Constitution has been controversial and widely debated since its ratification in 1866. The amendment states that “no State shall make or enforce any law which shall abridge the privileges or immunities of citizens of the United States,” although its imprecise definition leaves room for wide interpretations of what constitutes a citizen of the United States of America, as well as what rights these citizens are entitled to. [61] The issue of corporate rights has become more prevalent in conjunction with the rise of American capitalism. Corporations are not mentioned specifically in the Constitution; however, interpretations of the Fourteenth Amendment by the Supreme Court have granted corporations many of the same rights as citizens.
The first U.S. Supreme Court case to address corporate rights was Bank of the United States v. Deveaux in 1809, in which the Supreme Court ruled that corporations had the right to sue in federal court on behalf of citizens, but specified that corporations were not citizens. [62] In 1886, the Supreme Court continued this trend by stating that corporations were granted the same rights and protections under the Constitution as citizens in Santa Clara County v. Southern Pacific R. Co. [63] Almost a century later, the Supreme Court ruled in First National Bank of Boston v. Bellotti that corporate donations to ballot initiative campaigns were allowed as expressions of free speech under the First Amendment. [64] Bellotti was further upheld in Citizens United v. Federal Election Commission in 2010. [65] These rulings, along with many others, demonstrate how the classification of corporations as citizens has developed over more than 200 years, to the point where corporations are now entitled to the same rights as any other US citizen. While it is clear that corporations should be entitled to certain rights and protections, granting them the same status as an individual citizen inherently grants corporations more power than the citizen due to the massive amounts of resources, manpower, and influence that they hold. Furthermore, granting corporations the freedom to contribute to political campaigns allows for undue corporate influence on the legislative system that only further exacerbates the divide between a citizen and a corporate citizen.
A. Corporate Citizenship as Human Rights
As the extent of corporations increases over time, they have become both legally and politically incorporated into government and the state, granting them even more power. Due to their enormous influence, corporations have transformed into an “exceptional form of citizen that is empowered through the routine application of exemptions from, and exceptions to, the normal rule of law.” [66] This has led to both economic and political domination by the shareholders and executives of corporations, since they themselves have been granted the right to protection from the government. The seemingly unchecked display of power brings into question the ethical responsibilities of corporations, as well as the coercive nature of the relationship between corporations and consumers.
This power dynamic has become increasingly apparent in the pharmaceutical industry. The United Nations legally defines health as a fundamental human right, and obligates that its members ensure “equal access to health care and health-related services provided by third parties…ensure that privatization of the health sector does not constitute a threat to the availability, accessibility, acceptability and quality of health facilities, goods and services…[and] control the marketing of medical equipment and medicines by third parties.” [67] However, an important distinction is that these obligations set forth by the United Nations only apply to its states. Non-state actors, such as pharmaceutical companies, are only subject to human rights responsibilities, and it falls to the government to ensure that human rights are being protected. A 2008 report to the United Nations attempted to outline these responsibilities, which include the need for transparency and accountability in the pharmaceutical industry as well as discouraging the practice of lobbying for intellectual property laws and decreased drug price regulation. However, it is incredibly difficult to prove that companies are actually violating their responsibilities or punish them for doing so, due to a lack of concrete guidelines and the undue influence that the pharmaceutical industry has on the United States government. [68]
B. The Pharmaceutical & Healthcare Industry
The pharmaceutical and healthcare industry spent a total of $4.7 billion from 1999 to 2018 on lobbying and campaign contributions, with much of this money going toward campaigns against drug price regulation. [69] In comparison, consumers spent $5.5 trillion on prescription drugs over the same time period, further illustrating how little companies need to sacrifice in exchange for practically guaranteed protection from the government. [70] While all citizens are guaranteed the right to vote by the Constitution, it is clear that money also plays a significant role in American democracy, as there is a 60% chance that a law supported by a corporation will pass, compared to the 30% chance that a law supported by the majority of the American population will pass. [71] This unequal distribution of political power ensures that consumers and consumer groups who have significantly less resources than large corporations are automatically disadvantaged when it comes to influencing policy and campaigning for their rights.
It comes as no surprise that many of the top political spenders include corporations such as Pfizer and Eli Lilly, both companies which have come under fire for promoting fraudulent medications as well as price gouging. [72] However, this lobbying is not just limited to companies looking to make a profit. The American Medical Association (AMA), whose mission statement includes promoting “the betterment of public health,” contributed over $462 million toward ensuring that drug prices would not be lowered. [73] While this may seem incongruous with what organizations such as the AMA are trying to achieve, the influence of the pharmaceutical industry over the healthcare industry and the Department of Health is not one that should be overlooked. Public health agencies such as the FDA and the National Institutes of Health (NIH) are not immune to pressure from drug companies, thus limiting the “independence of regulatory bodies” and jeopardizing “critical democratic functions designed to protect public health.” [74] One example of this is the FDA’s 2005 decision to continue to allow the sale of COX-2 inhibitors that had a risk of adverse cardiovascular events, with one brand of the medication having been recalled due to severe side effects in patients. [75] It was later revealed that 10 out of the 17 panelists who voted in favor of the drugs had financial ties with the drugs’ manufacturers. [76]
The freedom granted to corporations by the Supreme Court has allowed companies to unduly influence both the legislative and executive branches of the government, resulting in direct harm for the consumer through increasing drug prices as well as untrustworthy regulatory practices. Without proper guidance or administration from the government, this relationship will only continue to grow and threaten the fundamental human rights of U.S. citizens.
VII. Economic Evaluation
There are two key dimensions to an analysis of the federal government’s financial interactions with private industries: the current state of the industry and its effect on the population on a wider scope. It is important to recognize the complex and diverse nature of federal interactions in the context of healthcare, and the consequently limited availability of specific and unique literature regarding the relationship between the federal government and private companies, as well as their effects. This analysis will address the level of abundance of that information and also provide foresight into the future of federal-private sector engagement.
A. The State of the Industry
This analysis will cover two types of federal interactions with private entities: those that are beneficial for private entities and those that are detrimental. The status quo consists of the use of several enforcement mechanisms to ensure the integrity of the health care sector to find equilibrium between profit-maximizing policies for the private industry and access to affordable health care for the individual. Although most public discourse on the regulation of the pharmaceutical industry emphasizes restrictive policies like price controls, private industry can benefit from acquired regulation. Corruption is overlooked and oftentimes facilitated by the federal government through methods like lobbying, direct subsidies, and regulation of competitors. Unfortunately, regulatory policies that benefit private industry in this way undermine the government’s obligation of ensuring the best quality of life for the citizens. In order to further comprehend the extent to which private entities have benefitted from regulation, it is important to reflect on the history of two very important entities in the health care scene: the Food and Drug Administration and the American Medical Association.
The lack of transparency, oversight, and accountability within the relationship between the federal government and the private sector of the healthcare industry has had devastating impacts on the individual level. Despite holding the fate of the quality of life for hundreds of millions in its hands, the federal government has prioritized profit-driven objectives and thus turned a blind eye to those who are suffering as a consequence. As mentioned above, there is a lack of literature directly analyzing the effectiveness of coercive (and acquired) regulations placed by the government due to its complex nature. Thus, the perception of the American citizen is crucial in determining the consequences of historical standards.
Beneficial regulations to private industries, known as acquired regulation, stem from 1930s legislation which included the introduction of mandated prescription medications. In 1938, the FDA passed legislation with the objective of curtailing self-medication: the practice of an individual purchasing any medication they see necessary without the approval of a professional. [77] The changes the federal government made to the drug market were unprecedented, under the assumption that it was unreasonable to predict that the average American would understand the ingredients list of medications or take the medication as instructed. This analysis does not concern the ethics of this assumption, but rather the consequences of a lack of transparency and foundational stability for years to come. As a result of the restrictive nature of the prescription market newly created by the FDA, it became extremely difficult for new drugs to be introduced into the market because of new regulations. Over time, the nature of the pharmaceutical industry became more monopolistic due to the lack of rigorous competition, allowing high prices to be set and causing fewer patients to receive care. This is a prime example of how systemic regulations and a lack of federal transparency can have dire consequences for the individual.
Conversely, the federal government may not deserve all the blame. It holds an immense array of responsibilities related to the healthcare industry, far beyond the administrative role it most famously plays. The Institute of Medicine of the National Academies explicitly outlined the duties of the federal government and its obligations to the citizens in the realm of healthcare, listing “regulator; purchaser of care; provider of [healthcare] services; and sponsor of applied research, demonstrations, and education and training programs for [healthcare] professionals.” [78] There are, in fact, several factors defined by the government itself that are structured to ensure the highest quality of care possible. As the analysts from the Institute of Medicine conclude, as both the purchaser and provider of healthcare, the federal government could establish economic incentives to increase the quality of care. For example, through financial bonuses and rewarding providers that achieve a higher level of care, the incentive to produce higher level care will naturally increase. [79] Additionally, the role the government plays as a provider of healthcare allows for the unique ability to individually select providers to contract with, also economically incentivizing health care providers to increase the quality of care that they receive. [80]
The analysis conducted through the Institute of Medicine identified two ways in which the federal government has interacted with healthcare providers in a restrictive nature: standards of participation and external review processes. Firstly, the standards of participation outline the minimal requirements that providers must adhere to. For the patient, these standards assume low levels of competence and health and safety regulation compliance. For the private provider, the standards encompass a diverse range of requirements starting from basic sanitation requirements to medical records management. [81] Moreover, the standards apply somewhat consistently throughout each state under a federally recognized regulation, with specific enforcement at the state level. Specifically, the enforcement capabilities are further outlined in a dual-leveled mechanism that places enforcement responsibilities under the Joint Commission on Accreditation of Healthcare Organizations. In order to ensure quality compliance, the Centers for Medicare and Medicaid Services are tasked with authoritative oversight. [82] Unfortunately, the effect of these policies as well as their long-term impact has not yet been evaluated, making these findings extremely difficult to apply in an active, modern day setting. The only way to identify any kind of relationship between regulation and quality of care is to analyze the trend of quality over time. The second method of coercive regulation is through external review, the process of third-party evaluation designed to maintain a high level of accountability and enforcement. [83] By adding an extra layer of evaluation through unbiased contracting, the government hopes to increase their level of transparency and ultimately improve the quality of care. However, due to a lack of consistency of standards between states and data collection, the accuracy of these external reviews faces a very big obstacle.
B. A COVID-Present Industry
In a COVID-19 context, the nature of the government’s role in pharmaceutical innovation has changed dramatically. Primarily, the role of the pharmaceutical industry in funding research and development has substantially decreased, while the government’s has increased. With the assistance of philanthropic organizations, government agencies have been a major contributor to the funding of not only research but also late-stage product development, increased manufacturing capacities, and efficient distribution of these products. [84] Fortunately, it allows for policies to be directly implemented without third-party intervention as well as increased enforcement and facilitation of those policies. This consequently allows for more humanitarian-focused objectives to be prioritized. The Coalition for Epidemic Preparedness Innovations, an organization that consists of both government agencies and non-profit organizations, proposed a multifaceted financing solution that sets costs only at the price of production aimed at lowering costs for the individual. [85] This optimistic legislation could potentially have very beneficial effects for the long term.
Despite dramatic changes in the control of financing and price-setting for the vaccine and medicine industry, it is unknown whether these changes will still be in effect after the pandemic ends. This creates greater uncertainty about the effectiveness of government subsidies in the long term, as well as the economic status of the pharmaceutical industry. In order for treatments for orphan conditions and therapies unrelated to the pandemic to become more affordable for individuals, the government’s financial role will remain a determining factor in long-term change.
VIII. Conclusion
In this article, we have attempted to provide a broad analysis of the regulatory framework governing United States food and drug safety. With particular attention to drugs like OxyContin and tobacco, the pharmaceutical industry at large, and regulatory agencies like the FDA, this article examines the power dynamics within American food and drug safety regulation. The above discussion of corporate rights is telling; an uneven power balance skewed toward private corporations dominates this landscape. We advocate for greater regulatory authority to be granted to agencies like the FDA, along with heightened measures to ensure agency accountability.
Case Law
Bank of the United States v. Deveaux, 9 U.S. 61 (1809)
Citizens United v. Federal Election Commission, 558 U.S. 310 (2010)
First National Bank of Boston v. Bellotti, 435 U.S. 765 (1978)
FDA v. Brown & Williamson Tobacco Corp., 529 U.S. 120 (2000)
Santa Clara County v. Southern Pacific Railroad Company, 118 U.S. 394 (1886)
Sottera, Inc. v. Food Drug Admin., 627 F.3d 891 (D.C. Cir. 2010)
United States v. Philip Morris USA Inc., 9F. Supp. 2d 1 (D.D.C. 2006)
U.S. v. Purdue Frederick Co., Inc., 495 F. Supp. 2d 569 (W.D. Va. 2013)
Congressional Bills
Affordable Insulin Now Act, H.R. 6833, 117th Congress 2799A-11 (2022)
Cosmetic Safety Enhancement Act of 209, H.R. 5279, 116th Congress (2019-2020)
Federal Food, Drug, and Cosmetic Act, 21 C.F.R. §§ 301-392 (1938)
Public Act 101-0625, I.L.G.A. SB0067, 101st General Assembly 356z.41 (2020)
Works Cited
[1] Sinclair, U. (1985). The Jungle. New York: Penguin Books.
[2] Borchers, A. T., Hagie, F., Keen, C. L., & Gershwin, M. E. (2007). The History and Contemporary Challenges of the US Food and Drug Administration. Clinical Therapeutics, 29(1), 1-16.
[3] Walker, D. M. 2007. Federal Oversight of Food Safety. GAO 08-435T. Washington, D. GAO.
[4] U.S. Department of Health and Human Services. (2011). FDA Mission.
[5] U.S. Chamber Institute for Legal Reform. (2013). The New Lawsuit Ecosystem.
[6] Felter, C. (2021). What Is the FDA’s Role in Public Health? Council on Foreign Relations. https://www.cfr.org/backgrounder/what-fdas-role-public-health.
[7] Griffin III, O. H., & Spillane, J. F. (2013). Pharmaceutical Regulation Failures and Changes: Lessons Learned from OxyContin Abuse and Diversion. Journal of Drug Issues, 43(2), 164-175.
[8] Griffin, O. H., & Miller, B. L. (2011). OxyContin and a Regulation Deficiency of the Pharmaceutical Industry: Rethinking State-Corporate Crime. Critical Criminology, 19(3), 213-226.
[9] U.S. v. Purdue Frederick Co., Inc., 495 F. Supp. 2d 569 (W.D. Va. 2013).
[10] Griffin, supra note 6.
[11] Food and Drug Administration. (2017). Frequently Asked Questions about the FDA Drug Approval Process.
[12] University of California San Francisco Academic Senate. (1994 ). Tobacco CEO’s Statement to Congress 1994 News Clip “Nicotine is not addictive.”
[13] U.S. Food and Drug Administration. (2018). Milestones in U.S. Food and Drug Law.
[14] FDA v. Brown & Williamson Tobacco Corp., 529 U.S. 120 (2000).
[15] United States v. Philip Morris USA Inc., 9F. Supp. 2d 1 (D.D.C. 2006).
[16] U.S. Food and Drug Administration. (2020). Family Smoking Prevention and Tobacco Control Act – An Overview.
[17] Sottera, Inc. v. Food Drug Admin., 627 F.3d 891 (D.C. Cir. 2010).
[18] Beran, D., Hirsch, I. B., & Yudkin, J. S. (2018). Why Are We Failing to Address the Issue of Access to Insulin? A National and Global Perspective. Diabetes Care, 41(6), 1125-1131.
[19] U.S. Food and Drug Administration. (2022). FDA Drug Competition Action Plan.
[20] Abernethy, A., & Woodcock, J. (2020). Insulin Gains New Pathway to Increased Competition, FDA.
[21] Public Act 101-0625, I.L.G.A. SB0067, 101st Gen. Assy. 356z.41 (2020).
[22] Affordable Insulin Now Act, H.R. 6833, 117th Cong. 2799A-11 (2022).
[23] Griffin, supra note 6.
[24] White, C.M. (2021). Why is the FDA Funded in Part by the Companies It Regulates? UConn Today.
[25] Medicaid. (2022). Medicaid & CHIP HOW-TO Information.
[26] Medicare. (2022). What’s Medicare?
[27] Ridic, G., Gleason, S., & Ridic, O. (2012). Comparisons of Health Care Systems in the United States, Germany and Canada. Materia Socio-Medica, 24(2), 112.
[28] Germany Visa. (2022). Health Insurance in Germany – The German Healthcare System.
[29] Id.
[30] Examining the Current State of Cosmetics: Hearings before the House Energy and Commerce Subcommittee on Health, 112th Cong., 2d (2012). (Testimony of Michael DiBartolomeis, CIH, Chief Occupational Lead, Poisoning Prevention Program and California, Safe Cosmetics Program, California Department of Public Health). https://www.govinfo.gov/content/pkg/CHRG-112hhrg78079/pdf/CHRG-112hhrg78079.pdf.
[31] Id, at 144.
[32] Federal Food, Drug, and Cosmetic Act, 21 C.F.R. §§ 301-392 (1938). https://www.govinfo.gov/content/pkg/COMPS-973/pdf/COMPS-973.pdf.
[33] U.S. Food and Drug Administration. (2021). FDA Authority Over Cosmetics: How Cosmetics Are Not FDA-Approved, but Are FDA-Regulated.
[34] Stoiber, T., Fitzgerald, S., & Leiba, N. S. (2020). Asbestos Contamination in Talc-Based Cosmetics: An Invisible Cancer Risk. Environmental Health Insights, 14, 1178630220976558.
[35] Id.
[36] Id.
[37] Compton, K. (2021). Johnson & Johnson (E. Miller, Ed.). Drugwatch. https://www.drugwatch.com/manufacturers/johnson-and-johnson.
[38] Id.
[39] Girion, L., & O’Donnell, C. (2019). J&J CEO Spurns U.S. Congressional Hearing on Carcinogens in Talc Products. Reuters. https://www.reuters.com/article/us-johnson-johnson-talc-congress/jj-ceo-spurns-u-s-congressional-hearing-on-carcinogens-in-talc-products-idUAKBN1YD2CA.
[40] Supra, note 31.
[41] Janetos, T. M., Akintilo, L., & Xu, S. (2018). Overview of High-Risk Food and Drug Administration Recalls for Cosmetics and Personal Care Products from 2002 to 2016. Journal of Cosmetic Dermatology, 18(5), 1361-1365.
[42] Id, at 1362.
[43] Id.
[44] Id.
[45] Id.
[46] Supra, note 28.
[47] Supra, note 28, at 144.
[48] Drabiak, K. (2017). Dying to Be Fresh and Clean? Toxicants in Personal Care Products, the Impact on Cancer Risk, and Epigenetic Damage. Pace Environmental Law Review, 35(1), 75-107.
[49] Id, at 75.
[50] Id, at 83.
[51] Id.
[52] H.R.5279 – 116th Congress (2019-2020): Cosmetic Safety Enhancement Act of 2019. (2020, March 11). https://www.congress.gov/bill/116th-congress/house-bill/5279.
[53] Supra, note 43.
[54] Wallack, G. (2019). Rethinking FDA’S Regulation of Cosmetics. Harvard Journal on Legislation, 56, 311.
[55] Id, at 332.
[56] Ruiz Cairo, E. (2015). Better Safe Than Sorry? The Impact of the EU-US Negotiations under TTIP on the Regulation of Cosmetic Products. Croatian Yearbook of European Law & Policy, 11(1), 115-132.
[57] Supra, note 49, at 332.
[58] Id.
[59] Id.
[60] Supra, note 28.
[61] U.S. Const. amend. XIV, § 1.
[62] Bank of the United States v. Deveaux, 9 U.S. 61 (1809).
[63] Santa Clara County v. Southern Pacific Railroad Company, 118 U.S. 394 (1886).
[64] First National Bank of Boston v. Bellotti, 435 U.S. 765 (1978).
[65] Citizens United v. Federal Election Commission, 558 U.S. 310 (2010).
[66] Whyte, D. (2018). The Corporate Citizen and the Sovereign Exception: From Homo Sacer to Homo Supra. Oñati Socio-Legal Series, 8(6).
[67] UN Committee on Economic, Social and Cultural Rights (CESCR). (2000). “General Comment No. 14: The Right to the Highest Attainable Standard of Health (Art. 12 of the Covenant).”
[68] Gruskin, S., & Raad, Z. (2010). Are Drug Companies Living Up To Their Human Rights Responsibilities? Moving Toward Assessment. PLoS Medicine, 7(9), e1000310.
[69] Wouters, O. J. (2020). Lobbying Expenditures and Campaign Contributions by the Pharmaceutical and Health Product Industry in the United States, 1999-2018. JAMA Internal Medicine, 180(5), 688-697.
[70] Id.
[71] Kornbluth, J., & Gilman, S. (Directors). (2017). Saving Capitalism.
[72] Supra, note 68.
[73] Id.
[74] Brezis, M. (2008). Big Pharma and Health Care: Unsolvable Conflict of Interests Between Private Enterprise and Public Health. Israel Journal of Psychiatry and Related Sciences, 45(2), 83.
[75] U.S. Food and Drug Administration. (2005). Analysis and Recommendations for Agency Action Regarding Nonsteroidal Anti-Inflammatory Drugs and Cardiovascular Risk.
[76] Supra, note 73.
[77] Temin, P. (1979). The Origin of Compulsory Drug Prescriptions. The Journal of Law and Economics, 22(1), 91-105.
[78] Smith, B. M., Eden, J., & Corrigan, J. M. (Eds.). (2003). Leadership by Example: Coordinating Government Roles in Improving Health Care Quality. National Academies Press.
[79] Id, at 58.
[80] Id, at 59.
[81] Id, at 61.
[82] Id, at 65.
[83] Id, at 67.
[84] Robinson, J. C. (2021). Funding of Pharmaceutical Innovation During and After the COVID-19 Pandemic. JAMA, 325(9), 825-826.
[85] COVAX. (2020). “COVAX Facility Explainer: Participation Arrangements for Self-Financing Economies.”